
Part 1: What is Type-1 Diabetes?
This article was developed and published in September 2018. Some of the views and evidence presented in this article may have evolved since their original development due to advancing literature and knowledge. Please look out for more recent articles, publications, etc... for the most up to date information revolving these topics.
I was diagnosed with Type-1 Diabetes on a family trip to Washington D.C. in 2007 when I was 16 years old. I remembered spending a week in the hospital learning about my new diagnosis. However, at that time, I did not appreciate the advancements and discoveries that had been made to make it possible for me to live years, not months, post-diagnosis. Speaking with Type-1 Diabetics who have lived with this disease for more than 50 years is an eye-opening experience. Stories of sterilizing old syringes for reuse in a boiling pot, no commercially available blood glucose meters until 1971, limited insulin options, etc. Now, Type-1 Diabetics can live much fuller, functional lives. However, a century ago, this was essentially a fatal diagnosis. In this Part 1 I will address:
Historical Perspective of Type-1 Diabetes
Biology of Type-1 Diabetes Initiation
Function Role of Insulin Producing Pancreatic Beta Cells
Pros and Cons of Having Type-1 Diabetes as a Patient and Researcher
Difficulties of Managing Type-1 Diabetes
Historical Perspective
Diabetes is a disease revolving around insulin. Currently, we now recognize that there are two distinct forms (insulin-deficient and insulin-resistant) of this disease and they are now separated into two categories (Type-1 and Type-2 Diabetes). Interestingly, this was not always clearly defined or accepted by the scientific community. Even into the 20th century, it was dogma that all diabetics were the same despite observations by scientists and physicians documenting what appeared to be separate “types” of diabetes.
Over 150 years ago, in 1866, a British physician was quoted as saying, “there are at least two distinct forms of the disease requiring diametrically opposing forms of treatment”(Harley, 1866). A decade later in 1877, Lancereaux, another well-known physician, distinguished two diabetic phenotypes, “thin” and “fat” diabetes (Lancereaux, 1880). Two other factors distinguished these “types” of diabetes at the time, age of onset and survival time after diagnosis. Type-1 Diabetes did, and still does, disproportionately strike those under the age of 18 years old (adult onset Type-1 is a minor fraction of the Type-1 Diabetic population, but is now becoming more common) (Maahs, West, Lawrence, & Mayer-Davis, 2010). Additionally, prior to the development of exogenous injectable insulin, Type-1 Diabetics typically didn’t survive longer than a few months post-diagnosis, whereas Type-2 Diabetics were able to live years post-diagnosis (Gale, 2001).
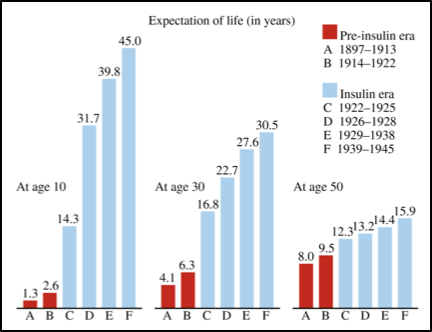
Figure 1. Life expectancy in pre- vs post insulin era. Figure 1 from (Gale, 2009). Gale illustrated the expected survival estimated by (Dublin, 1951). However, Gale went on to describe how this figure greatly overestimated survival in Type-1 Diabetics.
As you can imagine, without insulin, survival post-diagnosis in Type-1 Diabetes was extremely short and therapy options prior to the 1920s were limited to starvation (Mazur, 2011). Children often only had months to live, but starvation would prolong survival for a short period until children died of either ketoacidosis, malnutrition or tuberculosis (prevalent at the time). However, between 1920-1922 the biggest breakthrough for diabetes management was discovered over multiple key experiments. This discovery also resulted in a Nobel Prize awarded to Frederick Banting and John Macleod: The isolation and successful utilization of insulin for the treatment of diabetes ("The Discovery of Insulin," 2018).

Figure 2. Dr. Frederick Banting in his laboratory with one of his “subjects”
A summary of the experiments:
1) Removed the pancreas from dogs and blood sugar rose. Dogs experienced increased thirst, urination and fatigue, all cardinal symptoms of juvenile onset diabetes.
2) Reinjection of isolated pancreatic tissue into dogs (without pancreas) experiencing symptoms of insulin deficiency (hyperglycemia, ketoacidosis, etc.) restored health.
3) Isolation and purification of pancreas homogenates from larger animals (cow/bovine) to obtain larger quantities of pancreatic extract (insulin).
4) Banting and colleagues extracted and purified insulin, often injecting themselves to determine optimal insulin extract dose and hypoglycemia correction dosages with glucose rich foods (honey and orange juice).
5) Insulin injection into a near death 14-year-old diabetic patient restored health and reduced disease symptoms.
6) Numerous diabetics volunteered to receive pancreatic extract and experienced reduced disease burden.
For additional details on the discovery and controversy of insulin see: (Rosenfeld, 2002)

Figure 3. Dr. Sir Harold Himsworth’s 1936 discovery of different types of insulin metabolism in young vs old diabetes. Figure 3 from (Himsworth, 1940)
After the breakthrough discovery of insulin, in 1936, Dr. Sir Harold Himsworth developed an experiment which helped us distinguish the two types of diabetic patients (Himsworth, 1936). He took three patient populations: young diabetics, non-diabetics, and older diabetics. He administered BOTH oral glucose and injectable pancreatic extract (hereby referred to as insulin) into all patients. The young diabetics and non-diabetics both experienced hypoglycemic symptoms. However, the older diabetics did not experience hypoglycemic symptoms, but instead became hyperglycemic. This experiment clearly distinguished between these two biochemical forms of diabetes as insulin sensitive young diabetics vs. insulin resistance adult diabetics. The terms “Type-1” and “Type-2” diabetes arose around the same period from a population phenotyping technique popular in the early 20th century which defined groups by their shared characteristics and body type (called somatotyping) (Gale, 2001). To somatotype the “diabetic morphology,” Dupertuis examined 225 diabetic patients to understand whether diabetic were endomorphs, mesomorph, or ectomorphs (Draper G, 1940). To his and other’s surprise, he came to the discovery that there was not one, but instead TWO distinct morphological forms of diabetes. Consequently, they were referred to as Group I and Group II. Group I was lean, while Group II was typically obese. In 1951, Lister and colleagues expanded on this somatotyping work by combining it with the Himsworth experiment of insulin+glucose insulin sensitivity test (Lister et al., 1951). This experiment again elucidated two distinct groups, “Type-1” and “Type-2” based on morphology and insulin sensitivity/resistance. These results found that Type-1 diabetics had lean morphology and were insulin sensitive while Type-2 had obese morphology and insulin resistance. Today, this is STILL how we distinguish the two major forms of diabetes.

Figure 4. Lister et al. 1951 discovery combining both somatotyping and Himsworth insulin sensitivity (Himsworth, 1940). Figure 3 from (Lister, Nash, & Ledingham, 1951)
Read more on the history of Type-1 Diabetes here (Gale, 2001): http://diabetes.diabetesjournals.org/content/50/2/217.long
Type-1 Diabetes Initiation

Figure 5. Type-1 Diabetes Initiation. Immune cells auto-react to the insulin-producing beta cells found in the islet region of the pancreas.
Now, fast forward to the present…
Type-1 Diabetes is considered an autoimmune disorder where the insulin producing pancreatic beta-cells (or other component of the glucose regulatory process) are targeted and destroyed by our own immune system (Bluestone, Herold, & Eisenbarth, 2010). The environmental and genetic factors that drive this process are still not completely understood, but much of our current understanding of the pathophysiology of this disease is extrapolated from human patient observations and rodent modeling systems that mimic some of the human condition.
It is currently thought that the initiation of the Type-1 Diabetes autoimmune cascade starts with environmental immunological “insult(s)”. The “insult(s)” triggers the immune cells to look for pathogens within the body. However, inappropriately, these immune cells infiltrate the pancreatic islet cells and “tag” the insulin-producing beta-cells for immune system destruction, much like they would “tag” a pathogen (like bacteria, virus, etc). This “tag” will start a cascade of events which transmits a “kill” signal from one-immune cell to another, ultimately ending with T-cells (and other immune cells) now primed to react with the tagged insulin-producing beta-cells. This inappropriate immune reaction against your own tissues is where the term autoimmune comes from. Additionally, the body has a regulatory system in the Thymus (site of T-cell maturation) which prevents the production of auto-reactive T-cells, or T-cells that destroy your own body tissues. Thus, it is also thought that part of this thymic T-cell regulatory system becomes dysfunctional allowing for T-cells to react with host tissue. While Type-1 Diabetes isn’t considered a genetic disease, your genetics do contribute to how your immune system responds to pathogens and thus, Type-1 Diabetes has a “genetic” component (Concannon, Rich, & Nepom, 2009; Permutt, Wasson, & Cox, 2005). That said, genetics are not considered the primary driver of this disease, but a genetic history of Type-1 can slightly increase (5-10%) the susceptibility to developing the disorder (Redondo, Fain, & Eisenbarth, 2001).
The destruction of beta-cells is a slow process which typically doesn’t result in immediate destruction of all beta-cells, but lingers for weeks, months, and even years in some patients (Abdul-Rasoul, Habib, & Al-Khouly, 2006). This period of pancreatic destruction is termed the honeymoon period as some residual beta-cells still exist and produce insulin. However, for most Type-1 Diabetics, the majority of beta-cells will be destroyed within the year post-diagnosis, removing almost all previous blood glucose automation, requiring exogenous insulin injections to now control the disease.
A more detailed description of our current understanding of Type-1 Diabetes initiation is described in Bluestone et al. 2010 Nature publication (Bluestone et al., 2010).
What is the function of the Insulin Producing Pancreatic Beta Cells?

Figure 6. Normal beta-cell signaling in the presence of elevated blood glucose levels. 1) Glucose uptake via facilitated diffusion. 2) Glucose à ATP generation. 3) Closure of ATP-sensitive potassium channel. 4) Depolarize cell. 5) Ca2+ influx. 6) Exocytosis of insulin granules 7) Release of insulin into circulation. (Schuit, Huypens, Heimberg, & Pipeleers, 2001)
This automated process helps maintain normal blood glucose levels after meal ingestion. However, most Type-1 Diabetics have lost most or all their pancreatic beta-cell function. This means that a Type-1 Diabetic will not detect elevations in blood glucose and release insulin into their circulation. So, what happens when a Type-1 Diabetic has elevations in blood glucose? Well…when a Type-1 Diabetic ingests food that causes elevations in circulating blood glucose, the body has very few effective ways to remove this circulating glucose, causing glucose levels to remain elevated. This sustained elevation results in both acute and chronic damage to various tissues that will be discussed in more detail in later sections (Part 2).
The pancreatic beta cells have several critical functions, but one of the primary roles is to detect changes in blood glucose. These cells take glucose from the blood, through glucose transports (GLUT2), and converts it into ATP (Schuit et al., 2001; Thorens, Sarkar, Kaback, & Lodish, 1988). Elevated blood glucose, and subsequent elevations in ATP levels, initiate the rapid release of insulin from stored insulin granules directly into the circulation. This insulin will travel to various peripheral tissues, bind insulin receptors, and facilitate the translocation of glucose receptors (GLUT receptors) to the cell surface. These GLUT receptors (namely GLUT4 in the skeletal muscle, adipose, and cardiac tissue) allows for glucose to be co-transported into the cell with sodium. This causes circulating levels of glucose to be lowered by relocating glucose into peripheral tissues where it is typically stored and/or used for energy production (although not exclusively).
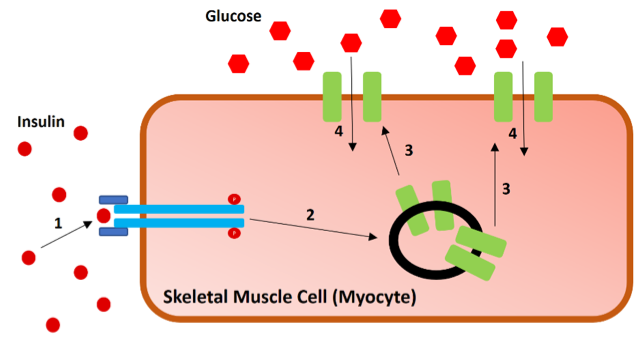
Figure 7. Insulin-induced glucose uptake into the skeletal muscle. 1) Insulin binds the insulin receptor (blue) causing the internal structure of the receptor to become phosphorylated. 2) Phosphorylated insulin receptor induces signaling cascade to GLUT4 (green) containing exosomes. 3) Exosomes transport GLUT4 receptors to the cell surface. 4) Glucose enters in through the GLUT4 transporters where it can be stored as glycogen, produce ATP, along with other cellular functions. See (Leto & Saltiel, 2012) for more details on insulin induce glucose uptake signaling. Ultimately, glucose levels within the blood begin to lower.
Non-insulin induced blood glucose lowering mechanisms:
While insulin facilitates the largest influence over glycemic control, the body does have another protective mechanism which can dispose of pathologically high blood glucose. While this is not as powerful, it is worth mentioning as it is currently the focus of new diabetic drug development (i.e. SGLT2 inhibitors). This mechanism includes glucose disposal through the urine and explains the hallmark symptoms of hyperglycemia: urination and thirst. Evidence indicates that each Type-1 Diabetic has a different concentration of glucose at which they begin to excrete glucose (Johansen, Svendsen, & Lorup, 1984). This threshold is termed the renal threshold for glucose and in a study of 23 Type-1 Diabetics, renal threshold for glucose ranged from 108-252mg/dL. SGLT2 inhibitors are now in study for Type-2 Diabetics. However, increased glucose within the urine has been linked to increased risk for urinary tract infections (Filippas-Ntekouan, Filippatos, & Elisaf, 2018).
Insulin’s primary mechanistic role in blood glucose regulation is to bind insulin receptors on the cell surface and induce the insulin signaling cascade. To reiterate, insulin binds, it facilitates the translocation of GLUT4 transporters to the skeletal muscle, adipose, and cardiac muscle tissue’s cell surface. However, insulin also works as a metabolic switch (at least acutely) where it can decrease gluconeogenesis, glycogenolysis, lipolysis, ketogenesis, and proteolysis, while increasing glucose uptake, glycogen synthesis, adipogenesis, and protein synthesis. Since the main effect of insulin on GLUT transporters is in the skeletal muscle, cardiac muscle and adipose tissue, these tissues are the primary benefactor of insulin-induced glucose uptake. The reason insulin is more powerful than any other endogenous blood glucose lowering mechanism is simply due to the sheer size and energetic need of these tissues (particularly the skeletal muscle). Additionally, the skeletal muscle is also the primary storage compartment for glucose [contains more glycogen than the liver (Jeremy M Berg, 2002)].
While the body does have other ways to modulate glucose levels, insulin-independent mechanisms pale in comparison to the powerful effect insulin has on regulating blood glucose levels. Simply put, without insulin the body cannot manage blood glucose levels automatically, forcing Type-1 Diabetics to use medicine (mostly in the form of exogenous insulin) to manually regulate blood glucose levels. However, absent the convenience of glycemic control automation in the form of pancreatic beta-cells, manually regulating blood glucose levels proves to be exceptionally difficult for most Type-1 Diabetics. In fact, the statistics on average blood glucose levels in Type-1 Diabetics: ARE. NOT. GOOD. I go into more details on Type-1 Diabetes management “success” in later Sections (Part 2).
Pros and Cons of Type-1 Diabetes as a Patient and Researcher
While having Type-1 Diabetes is no “cake walk”, it is certainly an incredibly insightful process. I often believe that having Type-1 Diabetes forces us (the diagnosed patient) on an incredible in-depth personal journey into the world of our own metabolism, how it works, how day-to-day life (sometimes moment-by-moment choices) influences it, and how these changes on metabolism can have far reaching effects over other aspects of our physiology. I often say that a professional athlete would kill for the level of personal insight Type-1 Diabetes can (often without choice) offer. Nutrition, exercise, sleep, circadian rhythm, lifestyle, medications, stress, etc., etc.… are all examples of factors that influence Type-1 Diabetes blood glucose and insulin management. Consequently, changes in one or more of these factors will alter insulin’s influence over glycemic control. The absence of insulin also gives insight into how the remaining functional parts of our metabolism regulate glucose in insulin’s absence. Additionally, Type-1 Diabetics are required (for disease management) to assess the biofeedback (insulin sensitivity, blood glucose responses, etc.) gathered from convenient Type 1 Diabetes glycemic management tools, getting constant insight into their own metabolism (again, whether they like it or not). Taken together, Type-1 Diabetics can manually regulate their own metabolism with insulin while simultaneously assessing how different factors such as lifestyle modulate these effects. From a researcher’s perspective, Type-1 Diabetics are the human model system for insulin knock-out. Consequently, Type-1 Diabetics are constantly experimenting and learning about how these factors influence our bodies. I find this to be one of the best assets I have as a researcher who studies metabolism. That said, I must also acknowledge that this is not an asset to all Type-1 Diabetics. Such a dynamic and information heavy diagnosis can create a substantial burden for many who are not interested in how, let’s say, “exercise intensity changes their insulin sensitivity”, but instead are just interested in living a normal burden-free and healthy life. However, for the self-experimenter who loves learning about their own metabolism, it is the ultimate gift. In fact, a lot of what we learn about in physiology and metabolism textbooks can be observed within the 24 hours of a Type-1 Diabetic’s day.
Why is Type-1 Diabetes so Difficult to Manage?

Figure 8 Representative 24-hour graph of a “normal” blood glucose curve for a healthy individual.
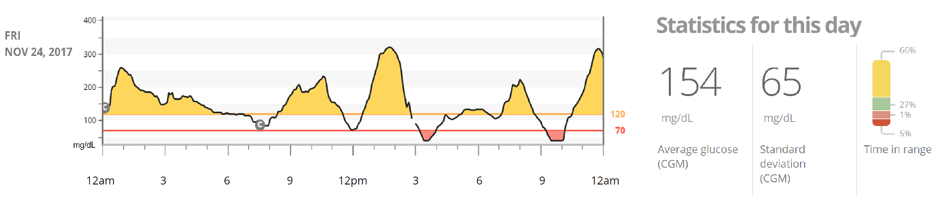
Figure 9 Representative 24-hour graph of a normal blood glucose curve for a Type-1 Diabetic.
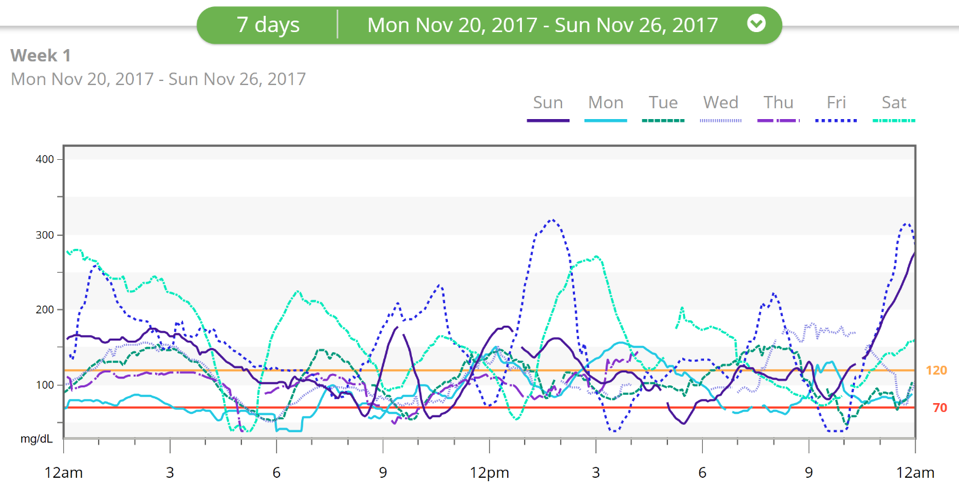
Figure 10 Representative weekly graph of daily blood glucose curves for a Type-1 Diabetic.

Figure 11 Factors influencing Type-1 Diabetic blood glucose standard deviation. Representative image of the standard deviation a Type-1 Diabetic may experience from the healthy individual’s normal blood glucose curve. Yellow lines represent deviation above normal blood glucose. Red lines represent deviation below normal blood glucose.
When viewing these different graphs (Figure 8-11), it QUICKLY becomes apparent that Type-1 Diabetics do not have the normal glycemic control of a normal healthy non-diabetic. In fact, they experience deranged blood glucose levels daily, which, if left uncontrolled, can result in damage in their body. This brings up the next question…
Why are Type-1 Diabetics often experiencing constant fluctuations in blood glucose levels weekly, daily, and often hourly?
Like much of our physiology (and even life), our metabolism is often in transition. For a Type-1 Diabetic, this presents a problem: any of the factors that influence blood glucose control are often in flux. Visualize a sinusoidal wave, going up and down. Unfortunately, that is an example of how our metabolism tends to work. It is not stagnant, but instead constantly changing based on a host on factors (many of which we have control over). Consequently, glycemic control truly requires a library of knowledge on how these dynamic factors influence blood glucose and insulin’s effectiveness, along with how the tools used by Type-1 Diabetics can be utilized to control blood glucose. I will describe some of these factors, though not an exhaustive list, and how they are thought to influence glycemic control.

Figure 12 Representative image of what goes into getting glycemic control. 1) Test blood glucose level and determine if it is high, low, or normal. 2) Determine the composition of your meal including grams of carbohydrates and protein, type of carbohydrates, etc. 3) Incorporate all confounding factors influencing glycemic control including exercise, sleep, stress, illness, time of day, caffeine, hormonal fluctuations, etc. 4) Take the information from your blood glucose and meal and determine how much insulin sensitivity will fluctuate due to this combination of lifestyle factors, which change daily. 5) Determine the type of insulin and quantity, and hope you get it right or you will experience hypoglycemia or hyperglycemia and require glucose or more insulin, respectively.
Daily factors influencing glycemic control:
· Sleep Quantity and Quality: Sleep is often seen as the rest and recovery portion of the day. It is ubiquitously appreciated that sleep has a tremendous influence on our health and the lack thereof can lead to adverse health outcomes. It has also been reported that reduced sleep quantity and/or quality can reduce insulin sensitivity. While the exact mechanism for reduced sleep induced insulin resistance is currently unknown, it is likely multifactorial (oxidative stress, inflammation, autonomic dysfunction, hormonal, amongst others).
Resource Link:
Arora and Taheri looked at the body of evidence on how sleep influences diabetes control but also insulin sensitivity (Arora & Taheri, 2015). They summarized 21 different studies on this topic in Table 5 with 19/21 studies indicating reduced insulin sensitivity (11-40%) with either reduced sleep quantity and/or quality: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4674464/
· Growth Hormone: Growth Hormone is often seen as metabolically antagonistic to insulin. Typically, in healthy individuals, when growth hormone is high, insulin is low. When insulin is low, growth hormone is high. However, we now appreciate that there is a role for endogenous (and exogenous) growth hormone production (and administration) in changing insulin sensitivity and glucose release (Perriello et al., 1990). This is most appreciated during periods of sleep. Usually in the later part of sleep, growth hormone elevates. When growth hormone elevates, insulin sensitivity begins to decrease and hepatic glucose output increases, playing two roles in increasing blood glucose levels. Mechanistically, this might be explained by growth hormone disrupting insulin signaling in adipose tissue (Takano et al., 2001).
Resource Links:
Growth Hormone Mechanism on Insulin Resistance: http://diabetes.diabetesjournals.org/content/50/8/1891
Review of Growth Hormone effect on Insulin Resistance: https://www.ncbi.nlm.nih.gov/pubmed/2406181
· Dawn Phenomenon: Dawn phenomenon refers to the elevation in blood glucose that accompanies the “dawn” period of day, when the sun begins to rise. It is currently believed that most individuals will see a 15-30mg/dL elevation in blood glucose during this period (Porcellati, Lucidi, Bolli, & Fanelli, 2013). While a lot is known about the dawn phenomenon, it is not completely understood. It is generally believed that growth hormone is a contributor to this temporal elevation in blood glucose (which also effects insulin sensitivity). However, adrenaline, cortisol, and glucagon are also relevant hormones which are known to elevate upon waking and are known to facilitate the glycogenolysis (breakdown of glycogen stores) and gluconeogenesis (creation of new glucose molecules from other molecules).
Resource Link: http://care.diabetesjournals.org/content/36/12/3860
· Exercise: Exercise is an important part of a healthy lifestyle. Exercise also plays a well-known role in altering insulin absorption, sensitivity, and binding potential, along with glucose output/release (Borghouts & Keizer, 2000). This also seems to depend on exercise type, intensity, and duration. While vital for health, exercise can introduce a powerful variable into glycemic control. https://www.ncbi.nlm.nih.gov/pubmed/10683091
Exercise…
o Increases insulin sensitivity: Anyone who exercises and has Type-1 (or Type-2) knows that exercise can increase the sensitivity of insulin (Borghouts & Keizer, 2000; Holloszy, 2005). There is not yet a consensus on the mechanism(s) of exercise-induced increases in insulin sensitivity, but emerging evidence indicates that AMPK (energy sensor within the cell that detects low “energy” levels) plays an important role in this process (Kjobsted et al., 2017).
https://www.ncbi.nlm.nih.gov/pubmed/16036907
https://www.ncbi.nlm.nih.gov/pubmed/27797909
o Increases absorption: Insulin is typically injected subcutaneously or intramuscularly. Exercise increases circulation rate and vasodilation. Thus, injected insulin absorption from the adipose tissue or skeletal muscle into circulation (where it now has access to all peripheral tissues) is expedited. While it does increase absorption of rapid-acting insulins [may be dependent on the injection site and exercise region (Ferrannini, Linde, & Faber, 1982)], it may not alter long-acting absorption rates (Berger, Cuppers, Hegner, Jorgens, & Berchtold, 1982; Peter et al., 2005). More on insulin type and kinetics in Part 3.
Other factors influencing insulin absorption rates: https://www.ncbi.nlm.nih.gov/books/NBK278938/table/insulin-pharmacology.factorcomm/
o Increases insulin binding potential: Exercise accompanies an elevation in heart rate. The elevation in heart rate means faster circulation of blood. Since insulin is within the blood, insulin will circulate much faster and thus have a great potential for encountering insulin receptors on peripheral tissues. Thus, increasing its binding potential.
o Increases insulin-independent glucose uptake in skeletal muscles: It is believed that repeated skeletal muscle contractions can increase the translocation of GLUT4 receptors to the skeletal muscle surface, independent of insulin. This means that glucose can be taken into the skeletal muscle cells, independent of insulin. That said, insulin still has the biggest influence on glucose uptake into peripheral tissues.
o Increase glucose output: Exercise can increase glucose output. This is a normal function of exercise to increase circulating glucose when it would be utilized by exercise. However, in Type-1 Diabetics, the mechanism for this may differ from normal healthy individuals. Petersen et al. 2010 found that the increase in glucose output during exercise could be entirely accounted for by increased gluconeogenesis [not glycogenolysis (Petersen, Price, & Bergeron, 2004)]. This was the opposite for healthy individuals.
· Nutrition: This plays the LARGEST role in glucose regulation and will require its own section (Part 3). This is the most important reason why Type-1 Diabetics need insulin, to shuttle the glucose produced from nutrient consumption to peripheral tissues for energy. Without food consumption, insulin requirements would be extremely low and would only be required to keep ketone and fatty acid metabolism in homeostasis (i.e. prevent diabetic ketoacidosis).
· Illness: Getting sick can be more serious for a Type-1 Diabetic. Illness often accompanies an increase in inflammatory status and stress molecules. Inflammation can increase insulin resistance, while stress molecules can increase glucose output. It is also problematic when a Type-1 Diabetic transitions out of their illness as insulin requirements can dramatically lower. Thus, going into and coming out of an illness can influence insulin sensitivity levels.
· Insulin Mismanagement: Insulin is the most powerful tool a Type-1 Diabetic has. It can be life-saving and, at times, life-threatening. Providing the right dose of insulin can often be akin to walking a tight rope due to the countless daily factors that influence its potency. Taking too much insulin can result in hypoglycemia. Taking too little insulin can result in hyperglycemia. Insulin mismanagement at one timepoint bleeds over into how you control later timepoints. Insulin management requires much further analysis and will be discussed in later sections (part 2 and 3).
· Diurnal Variation: Emerging evidence indicates that both our bodies and our cells run on an internal “clock” which alters physiology and signaling based on the time of day (circadian rhythm). Similarly, insulin sensitivity appears to change depending on the time of day (Hinshaw et al., 2013; Schrezenmeir J., 1993), although each individual’s sensitivity may depend on a myriad of lifestyle factors.
Resources:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3712033/
https://link.springer.com/chapter/10.1007/978-3-642-84855-1_7
· Other factors: Medications, Stress, …
What implications are there to having blood glucose levels that constantly fluctuate well above AND below the normal healthy range?
Are there consequences to blood glucose instability, are they just acute, or are there long-term complications and risks to these deranged blood glucose levels?
With a century and a half of research into this disease and massive advancement in medical technology, how much have Type-1 Diabetics benefited?
Can a Type-1 Diabetic expect to live the same life as a non-diabetic peer?
These questions and more will be addressed in Part 2 and 3 of this series
Key Messages:
Diabetes is a disease that revolves around insulin
Insulin was discovered through a series of experiments in the 1920’s and ultimately led to distinguishing between the two types of diabetes: Type-1 and Type-2
Type 1 Diabetes: typically early onset, insulin-deficient, but insulin sensitive (i.e. does not produce insulin but can respond to/utilize it)
Type 2 Diabetes: diagnoses typically later in life, insulin resistant (i.e. can produce insulin but does not respond to/utilize it)
The role of pancreatic beta cells is to detect changes in blood glucose and release stored insulin when blood glucose levels rise
The role of insulin it to regulate blood glucose by facilitating how glucose is taken into our cells from the blood stream (i.e. lower blood glucose)
Type-1 Diabetes is an autoimmune disorder where the body’s own immune system targets and destroys the insulin producing pancreatic beta-cells therefore no insulin can be produced
Type-1 diabetics rely on exogenous insulin to regulate blood glucose since their bodies don’t make it on their own
As a research who studies metabolism, Andrew has used his disease as a learning opportunity – through constant experimentation diabetes offers incredible insight into how the body works and how every factor in life can alter insulin’s influence over glycemic control (However, not the case for most Type-1 diabetics)
Type-1 Diabetes is incredibly difficult to manage due to the various different factors that influence blood glucose such as sleep quantity and quality, growth hormone, dawn phenomenon, exercise, nutrition, illness, insulin management, diurnal variation and many other factors
Even in a fasted state, the body produces residual glucose complicating management that much more
Due to the large range of variables that effect glycemic control, Type-1 Diabetics experience constant fluctuations in blood glucose
Stay tuned for Part 2 to understand the risks and complications of these blood glucose fluctuations and how current management strategies are attempting to offer glycemic control
References:
Abdul-Rasoul, M., Habib, H., & Al-Khouly, M. (2006). 'The honeymoon phase' in children with type 1 diabetes mellitus: frequency, duration, and influential factors. Pediatr Diabetes, 7(2), 101-107. doi:10.1111/j.1399-543X.2006.00155.x
Arora, T., & Taheri, S. (2015). Sleep Optimization and Diabetes Control: A Review of the Literature. Diabetes Ther, 6(4), 425-468. doi:10.1007/s13300-015-0141-z
Berger, M., Cuppers, H. J., Hegner, H., Jorgens, V., & Berchtold, P. (1982). Absorption kinetics and biologic effects of subcutaneously injected insulin preparations. Diabetes Care, 5(2), 77-91.
Bluestone, J. A., Herold, K., & Eisenbarth, G. (2010). Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature, 464(7293), 1293-1300.
Borghouts, L. B., & Keizer, H. A. (2000). Exercise and insulin sensitivity: a review. Int J Sports Med, 21(1), 1-12. doi:10.1055/s-2000-8847
Concannon, P., Rich, S. S., & Nepom, G. T. (2009). Genetics of type 1A diabetes. N Engl J Med, 360(16), 1646-1654. doi:10.1056/NEJMra0808284
The Discovery of Insulin. (2018). Retrieved from http://www.nobelprize.org/educational/medicine/insulin/discovery-insulin.html
Draper G, D. C., Caughey JL. (1940). The differentiation by constitutional methods between pancreatic diabetes and diabetes of pituitary origin. Trans Am Assoc Phys, 55, 146-153.
Dublin, L. I. (1951). The Facts of Life from Birth to Death. New York: MacMillan.
Ferrannini, E., Linde, B., & Faber, O. (1982). Effect of bicycle exercise on insulin absorption and subcutaneous blood flow in the normal subject. Clin Physiol, 2(1), 59-70.
Filippas-Ntekouan, S., Filippatos, T. D., & Elisaf, M. S. (2018). SGLT2 inhibitors: are they safe? Postgrad Med, 130(1), 72-82. doi:10.1080/00325481.2018.1394152
Gale, E. A. (2001). The discovery of type 1 diabetes. Diabetes, 50(2), 217-226.
Gale, E. A. (2009). How to survive diabetes. Diabetologia, 52(4), 559-567. doi:10.1007/s00125-009-1275-1
Harley, G. (1866). Diabetes, its various forms and different treatments. London: Walton and Mabberley.
Himsworth, H. P. (1936). Diabetes mellitus: its differentiation into insulin-sensitive and insulin-insensitive types. Lancet, i, 127-130.
Himsworth, H. P. (1940). Insulin Deficiency and Insulin Inefficiency. Br Med J, 1(4139), 719-722.
Hinshaw, L., Dalla Man, C., Nandy, D. K., Saad, A., Bharucha, A. E., Levine, J. A., . . . Basu, A. (2013). Diurnal pattern of insulin action in type 1 diabetes: implications for a closed-loop system. Diabetes, 62(7), 2223-2229. doi:10.2337/db12-1759
Holloszy, J. O. (2005). Exercise-induced increase in muscle insulin sensitivity. J Appl Physiol (1985), 99(1), 338-343. doi:10.1152/japplphysiol.00123.2005
Jeremy M Berg, J. L. T., Lubert Stryer. (2002). Biochemistry (5th ed.). New York: W H Freeman.
Johansen, K., Svendsen, P. A., & Lorup, B. (1984). Variations in renal threshold for glucose in Type 1 (insulin-dependent) diabetes mellitus. Diabetologia, 26(3), 180-182.
Kjobsted, R., Munk-Hansen, N., Birk, J. B., Foretz, M., Viollet, B., Bjornholm, M., . . . Wojtaszewski, J. F. (2017). Enhanced Muscle Insulin Sensitivity After Contraction/Exercise Is Mediated by AMPK. Diabetes, 66(3), 598-612. doi:10.2337/db16-0530
Lancereaux, E. (1880). Le diabete maigre: ses symptomes, son evolution, son prognostie et son traitement (Vol. 20). Paris.
Leto, D., & Saltiel, A. R. (2012). Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol, 13(6), 383-396. doi:10.1038/nrm3351
Lister, J., Nash, J., & Ledingham, U. (1951). Constitution and insulin sensitivity in diabetes mellitus. Br Med J, 1(4703), 376-379.
Maahs, D. M., West, N. A., Lawrence, J. M., & Mayer-Davis, E. J. (2010). Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am, 39(3), 481-497. doi:10.1016/j.ecl.2010.05.011
Mazur, A. (2011). Why were "starvation diets" promoted for diabetes in the pre-insulin period? Nutr J, 10, 23. doi:10.1186/1475-2891-10-23
Permutt, M. A., Wasson, J., & Cox, N. (2005). Genetic epidemiology of diabetes. J Clin Invest, 115(6), 1431-1439. doi:10.1172/JCI24758
Perriello, G., De Feo, P., Torlone, E., Fanelli, C., Santeusanio, F., Brunetti, P., & Bolli, G. B. (1990). Nocturnal spikes of growth hormone secretion cause the dawn phenomenon in type 1 (insulin-dependent) diabetes mellitus by decreasing hepatic (and extrahepatic) sensitivity to insulin in the absence of insulin waning. Diabetologia, 33(1), 52-59.
Peter, R., Luzio, S. D., Dunseath, G., Miles, A., Hare, B., Backx, K., . . . Owens, D. R. (2005). Effects of exercise on the absorption of insulin glargine in patients with type 1 diabetes. Diabetes Care, 28(3), 560-565.
Petersen, K. F., Price, T. B., & Bergeron, R. (2004). Regulation of net hepatic glycogenolysis and gluconeogenesis during exercise: impact of type 1 diabetes. J Clin Endocrinol Metab, 89(9), 4656-4664. doi:10.1210/jc.2004-0408
Porcellati, F., Lucidi, P., Bolli, G. B., & Fanelli, C. G. (2013). Thirty years of research on the dawn phenomenon: lessons to optimize blood glucose control in diabetes. Diabetes Care, 36(12), 3860-3862. doi:10.2337/dc13-2088
Redondo, M. J., Fain, P. R., & Eisenbarth, G. S. (2001). Genetics of type 1A diabetes. Recent Prog Horm Res, 56, 69-89.
Rosenfeld, L. (2002). Insulin: discovery and controversy. Clin Chem, 48(12), 2270-2288.
Schrezenmeir J., T. F., Tatò S., Laue C., Beyer J. (1993). Differences between Basal and Postprandial Circadian Variation of Insulin Sensitivity in Healthy Subjects and Type 1 Diabetics. Berlin, Heidelberg: Springer.
Schuit, F. C., Huypens, P., Heimberg, H., & Pipeleers, D. G. (2001). Glucose sensing in pancreatic beta-cells: a model for the study of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes, 50(1), 1-11.
Takano, A., Haruta, T., Iwata, M., Usui, I., Uno, T., Kawahara, J., . . . Kobayashi, M. (2001). Growth hormone induces cellular insulin resistance by uncoupling phosphatidylinositol 3-kinase and its downstream signals in 3T3-L1 adipocytes. Diabetes, 50(8), 1891-1900.
Thorens, B., Sarkar, H. K., Kaback, H. R., & Lodish, H. F. (1988). Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell, 55(2), 281-290.
